Chemo Resistance
Drug Sensitivity and Resistance in Cancer Chemotherapy
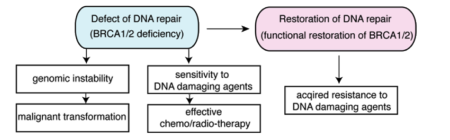
Defects in DNA repair lead to genomic instability and sensitivity to DNA damaging agents. Restoration of DNA repair in cancer cells can promote resistance to DNA damaging chemotherapeutic agents.
The Role of BRCA1/2 Reactivation in Resistance to Chemotherapy in BRCA1/2-Mutated Cancers
Defects in DNA repair result in increased genomic instability, which can lead to malignant transformation. Additionally, defects in DNA repair render cells sensitive to DNA damaging agents. BRCA1 and BRCA2 are important DNA repair proteins that are required for effective repair of DNA double-strand breaks by homologous recombination. Inherited mutations in BRCA1 and BRCA2 cause increased risk of developing various cancers, especially breast and ovarian cancers. Tumors that develop in patients with inherited BRCA1/2 mutations are generally believed to be BRCA1/2-deficient. Cancer cells with BRCA1/2 deficiency are defective in DNA repair by homologous recombination and sensitive to interstrand DNA crosslinking (ICL) agents, such as cisplatin and carboplatin, and poly(ADP-ribose) polymerase (PARP) inhibitors. Therefore, these agents are logical choices for the treatment for BRCA1/2-deficient tumors and are in fact clinically effective. However, acquired resistance to these drugs is a substantial obstacle for the treatment of BRCA1/2-mutated tumors.
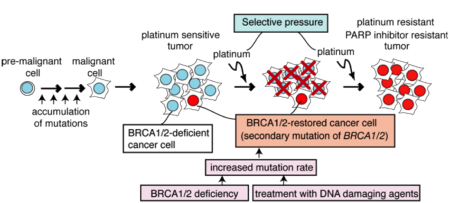
BRCA1/2 restoration due to BRCA1/2 secondary mutations is an important mechanism of cisplatin resistance in BRCA1/2-mutated tumors
Since BRCA1 and BRCA2 are critical factors for homologous recombination, which mediate repair of DNA damaged by cisplatin, we hypothesized that restoration of functional BRCA1/2 expression is one of the mechanisms of acquired cisplatin resistance of BRCA1/2-deficient cells. Indeed, we showed evidence for the restoration of BRCA1 and BRCA2 in platinum-resistant ovarian cancer samples obtained from BRCA1 and BRCA2 mutation carriers, respectively. We found secondary intragenic BRCA1/2 mutations that limited the severity of the initial mutation by restoring the transcript’s reading frame. Additionally, we carried out in vitro selection of BRCA2-mutated cancer cells in the presence of cisplatin and analyzed the surviving clones for BRCA2 protein expression. A large fraction of the cisplatin-selected clones re-expressed BRCA2 protein and were resistant to cisplatin. These clones acquired secondary mutations in the BRCA2 gene, either changes at the original mutation site or compensatory mutations close to the original mutation, which restored the reading frame. These clones with restored BRCA2 were cross-resistant to PARP inhibitor, a promising new drug which kills BRCA1/2-deficient cells effectively. These studies demonstrate that BRCA1/2 restoration is an important mechanism of cisplatin resistance and PARP inhibitor resistance. Since BRCA1/2 restoration does not explain all cases of cisplatin resistance, we are investigating other mechanisms of chemoresistance in BRCA-deficient cancer cells.